The NyuWa Chinese Population Variant Database (NCVD) is designed to collect single nucleotide polymorphisms (SNPs) and insertions or deletions (indels) in Chinese populations from high quality whole genome sequencing data, and to comprehensively annotate the variants on allele frequency in our Chinese dataset and external datasets (including 1KGP3, gnomAD), biology functions, phenotype, disease associations and pharmacogenomics.
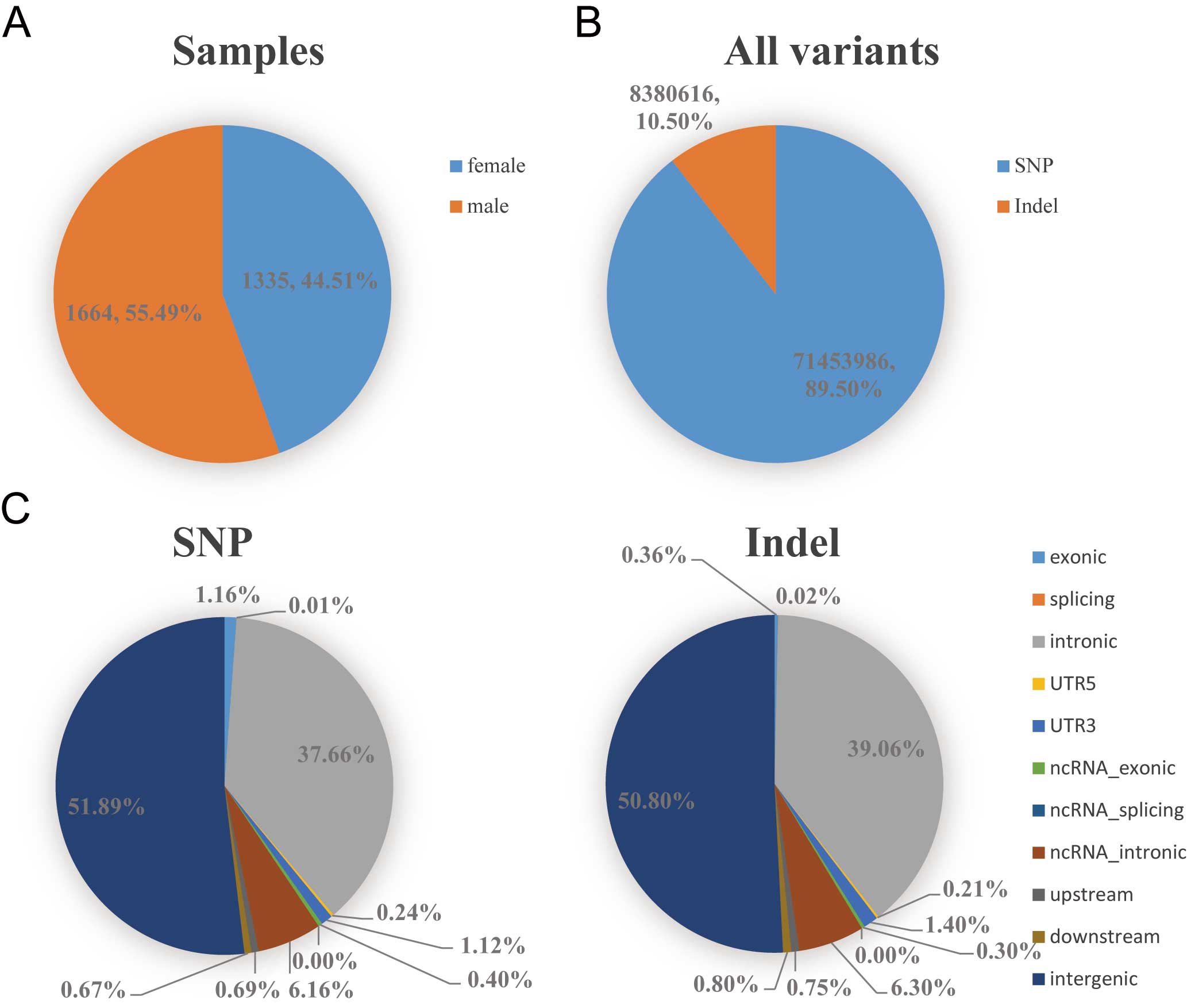
As its current version, NyuWa variants database archives 71 million (M) SNPs and 8 M indels based on high depth (median 26.2X) WGS of 2,999 Chinese individuals from 23 out of 34 administrative divisions in China. There are 32 M variants in protein coding gene region, including 857k CDS, 1.10M UTR, 8.60k splicing and 30.0M intron variants. As for functional protein coding variants there are 501k non-synonymous SNPs, 311k synonymous SNPs, 15.3k frameshift indels, 12.7k non-frameshift indels, 11.9k stopgains, 613 stoplosses.
Data analysis pipeline as follow.

The variant calling pipeline followed GATK [1] Best Practices Workflows Germline short variant discovery (SNPs + Indels) joint genotyping cohort mode. In brief, the raw sequencing reads were mapped to human reference genome assembly 38 with BWA-MEM v0.7.15 [2]. Picard (http://broadinstitute.github.io/picard/) was used to sort bam and mark duplicates. Mapping quality was check by qualimap v2.1.2 [3]. Indels were realigned and bases were recalibrated with GATK v 3.7. Variants were called for each sample using GATK HaplotypeCaller in ‘GVCF’ mode. GATK GenotypeGVCFs was then used to identify variants for all samples in the cohort. Then GATK VQSR was applied for SNPs and indels with truth sensitivity filter level 99.7 and 99.0, respectively.
The allele counts, allele frequency, homozygote number of variants are counted from dataset of 2999 Chinese individuals. The distribution of variants genotype quality and site quality are also counted. The genotype quality indicators include GQ, DP, allele balance for heterozygotes. The site quality indicators include FS, MQRankSum, InbreedingCoeff, ReadPosRankSum, VQSLOD, QD, DP, BaseQRankSum, MQ, ClippingRankSum.
The allele frequency from external datasets include population allele frequency from 1KGP3 [4] and the most recent released gnomAD version3.Variants were annotated with annovar v2018-04-16 [5] and Ensembl Variant Effect Predictor (ensembl-vep version 100.2) [6]. The adopted information from annovar comes from the following databases or tools: refGene, ensGene, avsnp150, clinvar_20190305, SIFT, Polyphen2. The adopted information from ensemble-vep comes from the plugin LOFTEE, which is developed recently by gnomAD to estimate the loss-of-function transcript effect [7].
The pharmacogenomics variants and related drug information were collected from 34 Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines. Then add the pharmacogenomics annotation to the variants in this database.
